
DNA僔乕働儞僒乕373A(Strech宆)
Key Word: DNA億儕儊儔乕僛/寀岝専弌/夋憸夝愅/僋儘儅僩僌儔儉/墫婎攝楍
偼偠傔偵
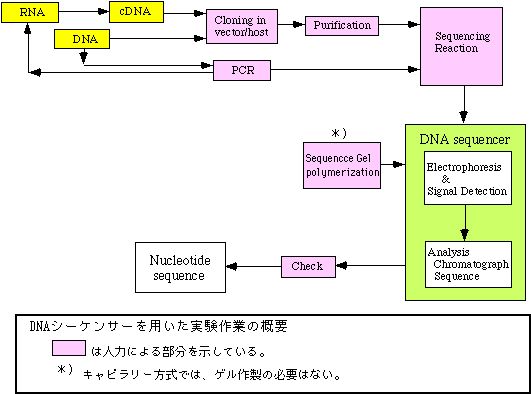
DNA偺墫婎攝楍寛掕媄弍偼丄堚揱巕偺峔憿婡擻傪尋媶偡傞偙偲偼傕偲傛傝恑壔宯摑偺尋媶傗屄懱幆暿娪掕傪巒傔條乆側栚揑偵梡偄傜傟傞斈梡媄弍偲側偭偰偄傞丅摿偵丄寀岝怓慺傪梡偄偰専弌偲攝楍暘愅偑帺摦壔偝傟偨僆乕僩儅僠僢僋DNA僔乕働儞僒乕偑奐敪巗斕偝傟偰埲棃丄墫婎攝楍寛掕偑挊偟偔岠棪壔偝傟偨偙偲傕偙偺媄弍偑峀偔晛媦偟偨梫場偲側偭偰偄傞丅杮復偱偼丄棟妛晹偱嫟摨棙梡偝傟偰偄傞DNA僔乕働儞僒乕373A(Strech宆)傪拞怱偵尨棟偲巊梡曽朄摍偵偮偄偰夝愢偡傞丅
栚師
______________________________
奐敪偺攚宨
尨棟偲惈擻
帋椏嶌惉偺嵽椏偺弨旛
帋椏嶌惉偺幚嵺
僎儖嶌惉偺梫揰
婡夿偺憖嶌
僨乕僞偺廋惓偲摑崌壔
堷梡暥專
______________________________
墫婎攝楍偺寛掕曽朄偑暘巕惗暔妛偺尋媶偵堦斒壔偟偨偺偼Maxam偲Gilbert偵傛傝壔妛揑尷掕暘夝曽朄乮Maxam仌Gilbert朄/1977擭丄暥專1乯偑妋棫偝傟偰埲棃偱偁傞丅Sanger摍偵傛傞DNA億儕儊儔乕僛偲僟僀僨僆僉僔僰僋儗僆僠僪傪梡偄傞曽朄乮僠僃乕儞僞乕儈僱乕僔儑儞朄偁傞偄偼僟僀僨僆僉僔朄/1977擭丄暥專2乯傕摨帪婜偵奐敪偝傟偨丅Maxam丄Gilbert偲Sanger偼偙偺岟愌偵傛傝僲乕儀儖壔妛徿傪庼梌偝傟偰偄傞丅Maxam仌Gilbert朄偼榑暥敪昞埲慜偵尋媶幰偺娫偵峀傑傝丄偙偺慜屻悢擭娫悽奅拞偱棙梡偝傟偨丅偦偺屻偼僟僀僨僆僉僔僰僋儗僆僠僪摍偺帋栻偑巗斕偝傟傞傛偆偵側偭偰偐傜丄僠僃乕儞僞乕儈僱乕僔儑儞朄偵傛傞寛掕曽朄偑庡棳偲側傝丄尰嵼偵帄偭偰偄傞丅
丂丂僠僃乕儞僞乕儈僱乕僔儑儞朄偼姰惉偝傟偨庤朄偱偁傞傕偺偺丄傾僀僜僩乕僾傪戝検偵梡偄丄斀墳乛揹婥塲摦暘棧乛専弌乛攝楍偺撉傒偲傝傑偱慡偰尋媶幰偵傛傞庤嶌嬈偱帪娫偲楯椡傪懡偔昁梫偲偡傞丅傑偨丄傾僀僜僩乕僾傪梡偄傞偨傔愱梡巤愝傪昁梫偲偟丄幚尡憖嶌拞偺旐敋乛墭愼傪忢偵峫椂偟偰幚尡偡傞昁梫偑偁偭偨丅1980擭戙偵傾僀僜僩乕僾偵戙傢偭偰寀岝怓慺傪梡偄傞曽朄偑峫埬偝傟偰丄偝傜偵専弌傪帺摦揑偵峴偆婡夿乮DNA僔乕働儞僒乕乯偑1986擭偵敪昞偝傟偨丅偦傟埲棃丄寀岝暔幙乛専弌曽幃/峺慺偺夵椙側偳偑奺幮偱嫞崌偟偰峴傢傟偨丅尰嵼偱偼傾僀僜僩乕僾朄傛傝傕斀墳傗婍嬶憖嶌偺柺偱傕娙曋偱惛搙傕崅偔側偭偨偨傔丄専弌傗攝楍撉傒庢傝偺帺摦壔偺儊儕僢僩偲憡傑偭偰丄峀偔DNA僔乕働儞僒乕偑棙梡偝傟偰偄傞丅
 丂
丂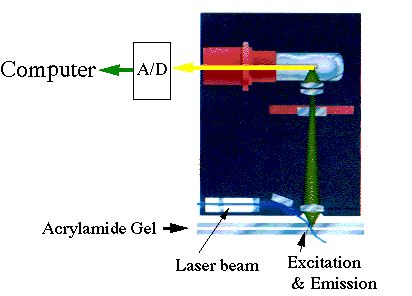
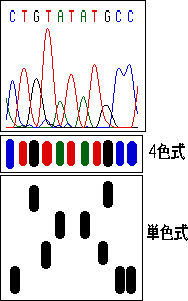
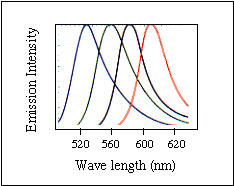
丂丂係庬椶偺寀岝暔幙傪摨帪偵梡偄傞曽幃(4怓幃丄暥專3乯傪傕偮PerkinElmer-ABI幮偺婡夿偲侾庬椶偺寀岝暔幙偺傒傪梡偄傞曽幃(扨怓幃丄暥專4乯偺Amersham-Pharmacia幮丄擔棫丄搰捗惢嶌強丄Aloka幮偺惢昳偑偁傞丅斀墳帋栻偺寀岝昗幆埵抲偱暘椶偡傞偲僆儕僑僰僋儗僆僠僪乮僾儔僀儅乕乯偵寀岝怓慺傪嫟桳寢崌偟偨傕偺傪梡偄傞曽幃乮僟僀僾儔僀儅乕朄乯偲僟僀僨僆僉僔僰僋儗僆僠僪乮僞乕儈僱乕僞乕乯偵寀岝怓慺傪嫟桳寢崌偟偨傕偺傪梡偄傞曽幃乮僟僀僞乕儈僱乕僞乕朄乯偑偁傞丅奺幮偺惢昳偲傕偙偺俀曽幃偵懳墳壜擻偲側偭偰偄傞丅
侾庬椶偺寀岝暔幙偺傒傪梡偄傞応崌乮扨怓幃乯偼丄傾僀僜僩乕僾傪梡偄偨僟僀僨僆僉僔朄偺応崌偲摨條偵侾偮偺僒儞僾儖DNA偵偮偄偰4庬偺斀墳傪暿乆偵峴偄丄係庬偺斀墳嶻暔傪偦傟偧傟暿屄偵揹婥塲摦暘棧専弌偡傞丅係儗乕儞偺僨乕僞傪侾偮偵傑偲傔偰侾偮偺僒儞僾儖DNA偺墫婎攝楍僨乕僞偵側傞丅帋栻丄専弌婡夿婡峔偑扨弮壔偱偒丄帋栻傕埨壙偱偡傓丅堦曽丄係庬椶偺寀岝暔幙梡偄傞応崌乮係怓幃乯偼丄帋栻丒憰抲偲傕崅壙偱偁傞偑丄扨怓幃偵斾傋偰斀墳憖嶌偑彮側偔偰偡傓偙偲偲丄揹婥塲摦偱侾僒儞僾儖摉偨傝1儗乕儞偟偐愯嫆偟側偄偺偱懡悢僒儞僾儖傪摨帪偵埖偊傞挿強偑偁傞丅
丂丂嫟摨棙梡偺僔乕働儞僒乕偼傾僋儕儖傾儈僪僎儖偵傛傞暘棧曽幃偱係怓寀岝帋栻傪梡偄嵟戝36屄偺僒儞僾儖傪摨帪偵埖偆偙偲偑偱偒傞丅寀岝専弌偼儗乕僓乕椼婲偺怓僼傿儖僞乕暘夝乮僶儞僪僷僗僼傿儖僞乕乯幃偲側偭偰偄傞丅Strech僞僀僾偵夵曄偝傟偰偄傞偨傔700-800墫婎挿傑偱偺暘棧擻傪帩偭偰偍傝丄暘棧擻偱偼嵟怴偺僔乕働儞僒乕偲摨摍偲側偭偰偄傞丅

帋椏嶌惉偵棙梡偝傟傞帋栻偼僉僢僩壔偝傟偰巗斕偝傟偰偄傞偺偱丄偙偙偱偼幚尡幰帺恎偱梡堄偟側偗傟偽側傜側偄DNA帋椏偲僾儔僀儅乕偵偮偄偰愢柧偡傞丅
DNA帋椏
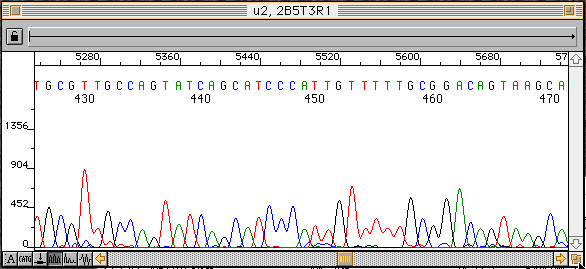
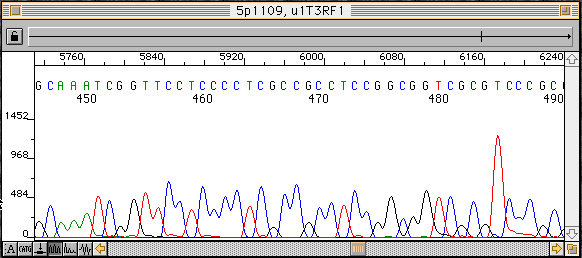
DNA僔乕働儞僒乕偺帋椏嶌惉偵梡偄傜傟傞僥儞僾儗乕僩DNA偺検
| DNA偺庬椶 | 僒僀僋儖僔乕働儞僗斀墳 | T7 polymerase斀墳 | ssDNA | 50-100ng | 200ng | Plasmid | 300-500ng | 5000ng | Cosmid, lambda Phage | 1000-2000ng | PCR Frgt. | 30ng- |
僾儔僀儅乕
儐僯僶乕僒儖僾儔僀儅乕偺棙梡丂pUC19側偳偺斈梡僾儔僗儈僪偱偼僋儘乕僯儞僌僒僀僩嬤朤偺攝楍傪帩偮僾儔僀儅乕偑巗斕偝傟偰偄傞丅M13-21, T7, T3僾儔僀儅乕側偳傛偔梡偄傜傟傞斈梡僾儔僀儅乕偼儐僯僶乕僒儖僾儔僀儅乕偲屇偽傟偰偄傞丅偙傟傜偼斈梡僾儔僗儈僪偵僋儘乕僯儞僌偝傟偨DNA偺攝楍傪撉傓偺偵棙梡偝傟偰偄傞丅儐僯僶乕僒儖僾儔僀儅乕偵寀岝暔幙傪晅壛偟偨傕偺傕巗斕偝傟偰偄傞偺偱僟僀僾儔僀儅乕朄偺斀墳偵傕棙梡偱偒傞丅儐僯僶乕僒儖僾儔僀儅乕傪梡偄傞応崌偵偼僋儘乕儞壔偝傟偨丂俢俶俙抐曅偺椉抂偐傜偦傟偧傟500墫婎掱搙偟偐攝楍傪撉傓偙偲偑偱偒側偄丅儐僯僶乕僒儖僾儔僀儅乕傪棙梡偟偰傛傝撪晹偺墫婎攝楍傪暘愅偡傞偨傔偵偼丄俢俶俙抐曅偺曅抂偐傜彮偟偢偮堎側偭偨挿偝偵寚幐傪嶌惉偟偨僾儔僗儈僪俢俶俙帋椏傪嶌傞昁梫偑偁傞乮僒僽僋儘乕僯儞僌朄丄僱僗僥僪僨儕乕僔儑儞朄側偳乯丅
僇僗僞儉僾儔僀儅乕偺棙梡丂墫婎攝楍傪撉傒偨偄晹埵偺50-200墫婎忋棳堟偺攝楍傪壔妛崌惉偟偰棙梡偡傞丅G+C娷検偑50亾掱搙偺応崌16墫婎挿偁傟偽傾僯乕儕儞僌壏搙俆侽搙偺斀墳偵棙梡偡傞偺偵廫暘偱偁傞丅嵟掅尷偵昁梫側僾儔僀儅乕偺梫審偼梈夝壏搙偺懠偵帋椏拞偵儐僯乕僋側攝楍偱偁傞偙偲偱偁傞丅僾儔僀儅乕撪偵憡曗揑側攝楍偑側偄丄挊偟偄墫婎偺曃傝偑側偄偙偲摍傕朷傑傟傞偑丄昁偢偟傕慡偰偺梫審傪枮偨偡攝楍傪梡偄傞偙偲偑偱偒側偄応崌偵昅幰摍偼俁乫抂偺嬤朤偱係庬偺墫婎偑傑傫傋傫側偔暘晍偟偰偄傞傕偺偱椙偄寢壥傪摼偰偄傞丅僾儔僀儅乕僨僓僀儞偺偨傔偺僜僼僩傕巗斕偝傟偰偄傞偺偱棙梡偝傟偨偄丅壔妛崌惉偼侾墫婎挿偁偨傝100-200墌偱嬈幰偵埶棅偱偒傞偑丄崌惉屻偺惛惢搙偵墳偠偰抣抜偼偝傜偵崅偔側傞丅DNA崌惉嬈幰偼僔乕働儞僗斀墳偵棙梡偡傞僆儕僑僰僋儗僆僠僪偲偟偰HPLC惛惢僌儗乕僪傪慐傔傞偑丄昅幰摍偼扙墫僌儗乕僪傪梡偄偰宱旓傪愡栺偟廫暘側寢壥傪摼偰偄傞丅
丂丂僟僀僾儔僀儅乕偺崌惉偍傛傃惛惢曽朄偵偮偄偰偼PerkinElmer-ABI幮偺僾儘僩僐乕儖傪嶲徠偝傟偨偄丅崌惉偼崅壙偱庤娫傕偐偐傞偺偱丄堦斒偺棙梡偼彮側偄偲峫偊傜傟傞偑丄儐僯僶乕僒儖僾儔僀儅乕傪棙梡偱偒側偄摿庩側儀僋僞乕傪梡偄偰懡悢偺僒儞僾儖傪夝愅偡傞傛偆側応崌偵偼桳岠偲巚傢傟傞丅
僠僃乕儞僞乕儈僱乕僔儑儞朄偱僞乕儈僱乕僞乕偲偟偰梡偄傜傟傞僟僀僨僆僉僔僰僋儗僆僠僪偼僨僆僉僔僰僋儗僆僠僪偲嫞崌娭學偵偁傝丄Taq億儕儊儔乕僛側偳偺懴擬惈DNA億儕儊儔乕僛偺斀墳偵庢傝崬傑傟偵偔偄丅寀岝怓慺傪曐帩偟偰偄傞僟僀僞乕儈僱乕僞乕偼丄僟僀僨僆僉僔僰僋儗僆僠僪偵斾傋偰偝傜偵懴擬惈DNA億儕儊儔乕僛偺斀墳偵庢傝崬傑傟偵偔偄偨傔丄僒僀僋儖僔乕働儞僗斀墳偼DNA僔乕働儞僒乕梡偺帋椏嶌惉朄偲偟偰偼晛媦偟側偐偭偨丅偟偐偟丄懴擬惈DNA億儕儊儔乕僛偺夵椙偑峴傢傟偰僟僀僞乕儈僱乕僞乕偺庢傝崬傒岠棪偑岦忋偟偨寢壥丄尰嵼偱偼嵟傕憖嶌偑扨弮偱彮検偺帋椏偱拝幚側寢壥傪弌偣傞斀墳朄偲側偭偰偄傞丅
PerkinElmer-ABI幮偺僉僢僩傪梡偄傞応崌偺斀墳椺傪埲壓偵婰嵹偡傞丅
((*)ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit)
憖嶌偑娙扨偱丄帋椏DNA検傕彮側偔偰偡傓偙偲偑夝傞丅
_______________________________________________________
| Terminator Ready Reaction Mix(*) | 8microL | Primer(3microM) | 1microL | Plasmid 梟塼(0.5microgram) | 11microL |
 屌壔拞偺僎儖
屌壔拞偺僎儖
僎儖嶌惉梡偺帋栻偼偱偒傞偩偗崅弮搙帋栻傪梡偄傞丅摿偵傾僋儕儖傾儈僪梟塼傗僂儗傾偼屆偄傕偺傪梡偄側偄丅傾僋儕儖傾儈僪偲僂儗傾偺梟塼傪僀僆儞岎姺庽帀偱張棟偡傞偙偲偼昅幰摍偼峴偭偰偄側偄偑丄廳崌壔怗攠偺揧壛偺慜偵尭埑扙婥傪寭偹偰僼傿儖僞乕鄅夁傪偟偰偄傞丅
僎儖偺屌傑傞僗僺乕僪傕梫場偲側傞丅僎儖梟塼拲擖帪偵婥朅偑偱偒傞偲拲擖傪拞抐偟側偗傟偽側傜側偔側傞丅拞抐傪峫椂偵擖傟偰丄婥朅傪捛偄弌偡偨傔偺備偲傝帪娫傪妋曐偡傞偨傔丄僎儖梟塼傪椻媝偡傞偲偄偆搘椡傪偡傞棙梡幰傕偄傞丅婥朅偺敪惗傪旔偗傞偨傔偵僈儔僗斅偼墭傟傗毢偺柍偄傛偆偵偟丄庢傝埖偄偵側傟偨恖偺憖嶌傪娤嶡偟偰廋摼偡傞丅傑偨丄僎儖偺屌壔懍搙偼幒壏傗僈儔僗斅偺壏搙偵埶懚偡傞丅昅幰摍偼廳崌壔怗攠偺偆偪夁棸巁傾儞儌僯僂儉塼偺検傪壞婜偼敿暘偵尭傜偟偰屌壔僗僺乕僪傪挷愡偟偰偄傞丅拲擖屻偺屌壔帪娫偼俀帪娫掱搙偱廫暘偩偲尵傢傟偰偍傝丄俆帪娫傎偳偺娫偵塲摦傪奐巒偟偰偄傞丅

僔僌僫儖偺専弌晹埵偼僎儖壓抂偐傜侾侽僙儞僠掱搙忋偺廲侾僙儞僠偺椞堟偱偁傞丅摿偵偙偺晹暘偵彎傗墭傟偑偮偐側偄傛偆偵僈儔僗斅傪庢傝埖偆丅偙偺晹暘廃曈埲奜偱偼僎儖斅偐傜僂儗傾傗僎儖曅傪偒傟偄偵彍嫀偱偒偰偄傟偽栤戣側偄丅僎儖屌壔屻偵憰抲杮懱傊憰拝偡傞慜偺傾儖僐乕儖偱偺怈偒庢傝偵嵺偟偰偼丄忋婰偺儗乕僓乕徠幩晹偺傒娙扨偵峴偆丅峀偄柺愊偵傢偨傝傆偒庢傠偆偲偡傞偲丄偐偊偭偰墭傟傪峀偘偰彎傪懡偔偮偗傞偙偲偵傕側傞偺偱丄拲堄偡傞昁梫偑偁傞丅
丂丂婡夿偺憖嶌偼愝抲偝傟偰偄傞愭抂婡婍偺拞偱偼嵟傕扨弮偱偁傝丄婛偵憖嶌偵側傟偨恖偲俀夞掱搙堦弿偵憖嶌傪偡傟偽廋摼偱偒傞丅埲崀偼儅僯儏傾儖傪尒側偑傜幚峴偱偒傞偺偱妶梡偝傟偨偄丅
丂丂嵟傕棙梡昿搙偺崅偄婡夿側偺偱棙梡帪娫傪庣傞偙偲偲僩儔僽儖偑婲偒偨偲偒偵懍傗偐偵懳張偡傞昁梫偑偁傞丅栭偵塣揮傪奐巒偟偰僩儔僽儖偑婲偙偭偨帪丄儊乕僇乕偵楢棈偟偰傕媄弍幰偼婛偵婣戭偟偰偄傞丅僩儔僽儖偺懳張偼梻擔埲崀偵側傝丄夝寛偑挿堷偔暘偩偗埲崀偺棙梡梊掕幰偵偟傢婑偣偝傟傞丅屵屻憗偔偵塣揮傪巒傔傞偺偑僩儔僽儖傪嵟彫嵟抁婜娫偵偡傞僐僣偱偁傞丅
婡夿偺憖嶌儊僯儏乕
1 婡夿偺梊栺偲塣揮偵昁梫側帪娫
2 婡夿憖嶌偺幚嵺
x 揹婥塲摦傪奐巒偡傞帪崗偑抶傟偨応崌
######################
1 婡夿偺梊栺偲塣揮偵昁梫側帪娫
######################
DNA僔乕働儞僒乕偺愭栺忬嫷偺妋擣偲棙梡梊栺偼on line偱偱偒傞丅
(cellvx.bio.sci.hiroshima-u.ac.jp)
棙梡梊栺傪偟偨擔偵棙梡偡傞丅
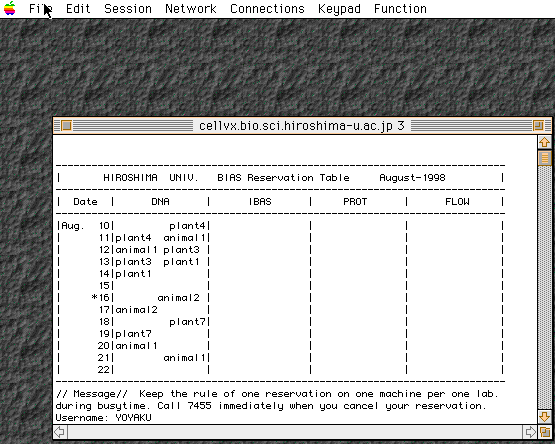
梊栺夋柺
杮憰抲偱儘儞僌僎儖傪梡偄傞偲丄婲摦偟偰偐傜僨乕僞偺報嶞傑偱偵栺俀侽帪娫偐偐傞丅
梻擔偺棙梡梊栺幰偑屵屻偐傜棙梡偱偒傞傛偆偵丄婡夿棙梡偼抶偔偲傕屵屻俆帪偵偼奐巒偡傞丅15帪娫偺僎儖揹婥塲摦偱800墫婎挿傑偱偺僔僌僫儖専弌偑峴傢傟傞丅儊乕僇乕偺儅僯儏傾儖偱偼侾俉帪娫偺塲摦傪慐傔偰偄傞偑丄15帪娫埲忋偵挿帪娫揹婥塲摦偟偰傕丄僔僌僫儖偼摼傜傟傞傕偺偺暘棧擻偺尷奅傪挻偊偰偄傞偺偱幚幙揑偵柍懯偲側傞偙偲偑懡偄丅婡夿偺棙梡傪屵屻4帪偵巒傔傟偽廔栭塣揮偟梻擔偺挬8帪崰偵揹婥塲摦偑廔椆偡傞丅帺摦夝愅偵侾帪娫掱搙丄報嶞偵侾-2帪娫偐偐傞丅儅僯儏傾儖儗乕儞僩儔僢僉儞僌傪挬9帪偐傜巒傔俀侽暘屻偵夝愅偲報嶞傪巒傔傞偲丄屵屻侾帪偵偼慡偰偺憖嶌偑廔椆偡傞丅
婡夿偺憖嶌儊僯儏乕傊栠傞
儊僯儏乕傊栠傞
######################
2 憖嶌偺幚嵺
######################
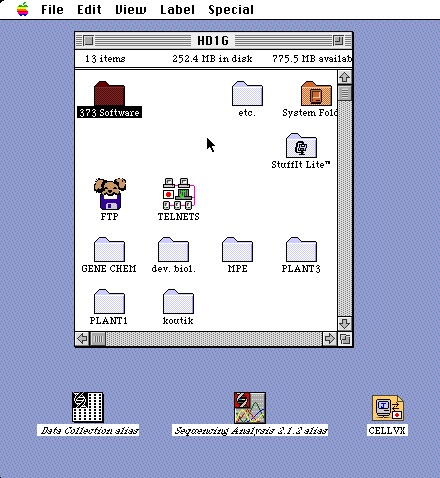
僐儞僺儏乕僞乕偺弶婜夋柺
憰抲杮懱偲僐儞僺儏乕僞偺揹尮傪擖傟傞丅

僐儞僺儏乕僞偱cellvx偺傾僀僐儞偱telnet愙懕偟丄儐乕僓乕柤+僷僗儚乕僪偱儘僌僀儞偡傞丅
4.DNA 偵偰DNA僔乕働儞僒乕傪棙梡奐巒偡傞偙偲傪楢棈偡傞丅
妋擣傪媮傔偰偔傞偺偱丄4+儕僞乕儞偡傞丅
棙梡奐巒愰尵
壽嬥奐巒偺昞帵偑偁傝丄帺摦揑偵儘僌僆僼偝傟傞丅
儊僯儏乕僶乕撪偺廔椆傪慖傃telnet僾儘僌儔儉傪廔椆偡傞丅
Data Collection偺傾僀僐儞偱婡夿杮懱偐傜偺僨乕僞庴怣壜擻側忬懺偵偡傞丅
埲忋偺憖嶌偺娫偵憰抲杮懱偼僙儖僼僠僃僢僋傪廔偊偰僨傿僗僾儗乕偵Main Menu偑昞帵偝傟傞丅
揹婥塲摦忦審傪愝掕偡傞丅乮儘儞僌僎儖偺応崌偼2800V 40mA 42W 15Hr乯
Main Menu偵栠傝丄PreRun儊僯儏乕偵堏傞丅
僎儖斅偺儗乕僓乕徠幩偝傟傞晹暘傪僀僜僾儘僷僲乕儖傪彮検偮偗偨巻偱寉偔怈偔丅
丂丂屌壔偟偨僎儖斅偼師偺嶌嬈傪嵪傑偣偰偍偔丅僥乕僾傪偼偢偟偰悈摴悈偱昞柺傪傛偔愻偄僂儗傾傗僎儖曅傪愻偄棊偲偟偰偍偔丅忲棷悈偐僀僆儞岎姺悈偱儕儞僗屻偵姡偐偡丅
 憰抲杮懱偵憰拝偝傟偨僎儖斅
憰抲杮懱偵憰拝偝傟偨僎儖斅
杮懱偺塲摦晹斷傪奐偒壓晹僶僢僼傽乕憛偲僎儖斅傪憰拝偟屌掕偡傞丅塲摦晹斷傪暵偠傞丅
PreRun儊僯儏乕偱Plate Check傪奐巒偡傞丅
僎儖偵寀岝専弌偺忈奞偲側傞傛偆側栤戣偑側偄偐専嵏偡傞偺偑Plate Check偱偁傞丅
僐儞僺儏乕僞夋柺偵僎儖斅偺憱嵏忬嫷偑昞帵偝傟傞丅憱嵏慄偵棎傟偑偁傟偽僎儖斅偵墭傟傗彎偑偁傞偺偱丄憰抲偐傜庢傝奜偟偰昞柺傪妋擣偟墭傟傪庢傞丅墭傟傗彎偑昞柺偱側偔撪晹偺応崌偼庢傝彍偗側偄偺偱丄偦偺埵抲偵憡摉偡傞儗乕儞偵僒儞僾儖傪嵹偣側偄傛偆埵抲傪妋擣偟偰偍偔丅埵抲偼夋柺忋偱憱嵏慄偺棎傟偨埵抲偵儅僂僗偺億僀儞僩傪抲偔偲偦偺嵗昗偑昞帵偝傟傞偺偱妋擣偱偒傞丅
忋晹僶僢僼傽乕憛偲僒儞僾儖僐乕儉傪憰拝偟丄TBE僶僢僼傽乕傪壛偊傞丅
楻揹傗僔儑乕僩側偳恟戝側僩儔僽儖偺尦偵側傞偺偱丄僶僢僼傽乕偑楻傟偨傜懍傗偐偵傆偒庢傞丅僶僢僼傽乕拲擖屻偵忋晹塲摦憛偲僎儖斅偲偺娫偐傜楻傟偑側偄偐傕僠僃僢僋偡傞丅
塲摦晹斷傪暵偠偰PreRun儊僯儏乕偱PreRun傪奐巒偡傞丅
杮懱偺昞帵晹偵幚揹埑偲揹棳偑昞帵偝傟傞偺偱昁偢悢抣傪妋擣偡傞丅
偙偙偱1000V埲忋壛埑偝傟偰偄側偗傟偽丄憖嶌傪拞抐偡傞丅
丂僎儖偍傛傃僶僢僼傽乕傕偟偔偼婡夿偺僩儔僽儖偑憐掕偝傟傞丅
PreRun帪揰偱偺妋擣偱僩儔僽儖傪抦傟偽丄帋椏埲慜偺栤戣傪枹慠偵夞旔偱偒丄崅壙側帋椏傪柍懯偵偟側偄偱偡傓丅

PreRun屻偵丄僒儞僾儖僂僃儖傪儕儞僗偟偰偐傜僒儞僾儖傪拲擖偡傞丅
儕儞僗偑晄廫暘偩偲僒儞僾儖偑棎傟偰僔僌僫儖偑僔儍乕僾偱側偔側傝暘棧擻偑壓偑傞丅
僒儞僾儖悢偑懡偄応崌偼婏悢儗乕儞偵傾僾儔僀偟偰侾侽暘娫塲摦偟丄嵞搙儕儞僗傪偟偰嬼悢儗乕儞偵傾僾儔僀偡傞丅Main Menu偐傜Run傪巜帵偟偰塲摦傪奐巒偱偒傞丅

僐儞僺儏乕僞懁偱偼丄儊僯儏乕僶乕偐傜setting傪慖傇丅
僨乕僞庢傝崬傒帪娫傗僒儞僾儖悢丄僾儔僀儅乕偺庬椶丄帺摦夝愅傗報嶞偵偮偄偰偺巜帵傪偡傞丅
儊僯儏乕僶乕偐傜New sample file嶌惉偵偼偄傞丅
僒儞僾儖僼傽僀儖偺柤慜傗僐儊儞僩丄夝愅傗報嶞偵偮偄偰偺屄暿偺巜帵偑偙偙偱偱偒傞丅
嵟屻偵丄杮懱偐傜憲怣偝傟偰偔傞僔僌僫儖傪僐儞僺儏乕僞乕偑僴乕僪僨傿僗僋偵曐懚偟偰偄傞偐妋擣偟偰丄僨傿僗僾儗僀傪埫偔偟廔栭塣揮偡傞丅曐懚傪巜帵偱偒偰偄側偄偲丄偄偭偝偄偺僨乕僞偼幐傢傟傞丅
梻挬丄杮懱偺僨傿僗僾儗僀偱偼Run Completed偲昞帵偝傟偰偄傞丅
搑拞偵僩儔僽儖偑偁傟偽僄儔乕儊僢僙乕僕偑偱傞偺偱昁偢儊儌偡傞丅
憰抲杮懱偺揹尮傪愗傞丅
僶僢僼傽乕丒塲摦憛偲僎儖斅傪庢傝奜偡丅
忋晹偺僶僢僼傽乕傪媯傒弌偟偰彮検偵偟偰偐傜丄僎儖斅偲忋晹僶僢僼傽乕憛偑崌懱偟偨傑傑杮懱偐傜庢傝奜偡偺偑丄僶僢僼傽乕傪杮懱偵楻傜偝側偄僐僣偱偁傞丅杮懱偵僶僢僼傽乕偑楻傟偰偄傟偽彮検偱傕昁偢傆偒庢傞丅僶僢僼傽乕偺楻傟偑専弌晹傗婡夿揹婥晹偵偍傛傇偲嵟埆偱偁傞丅偙偺塭嬁偼師夞偐傜偺棙梡幰偵弌尰偡傞丅
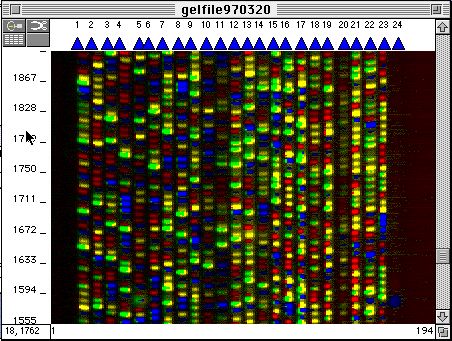 僎儖僀儊乕僕
僎儖僀儊乕僕僐儞僺儏乕僞懁偱偼丄揮憲偝傟偨僔僌僫儖傪僎儖僼傽僀儖偵挋傔偰丄偙傟傪婎偵僎儖僀儊乕僕傪嶌傞丅僎儖僀儊乕僕忋偺儔僟乕傪偨偳偭偰儗乕儞僩儔僢僉儞僌偝傟傞丅僒儞僾儖僼傽僀儖偼帺摦揑偵嶌傜傟傞丅帺摦夝愅傪巜帵偟偰偁傟偽丄Analysis僜僼僩偑婲摦偝傟偰奺僒儞僾儖偺夝愅偑巒傑傞丅
帺摦儗乕儞僩儔僢僉儞僌偼昁偢偟傕惓偟偔側偄丅摿偵丄嵍懁偵僒儞僾儖偑柍偄応崌傗僔僌僫儖偺庛偄僒儞僾儖偑懡偄偲丄僐儞僺儏乕僞偼儗乕儞傪妋幚偵尒岆傞丅彮側偔偲傕丄僎儖僀儊乕僕偺奣娤偐傜奺儗乕儞偵敀慄偑岆傝柍偄弴斣偲埵抲偱堷偐傟偰偄傞偐妋擣偡傞丅
儗乕儞僩儔僢僉儞僌傪庤摦偱峴偭偨応崌丄帺摦夝愅偲報嶞傪巜帵偡傞丅夝愅僨乕僞偼愭偢丄Raw僨乕僞偲側傝丄偙傟傪婎偵僋儘儅僩僌儔儉偲墫婎攝楍偑拪弌偝傟傞丅偄偢傟傕僒儞僾儖僼傽僀儖偵彂偒崬傑傟傞丅
僒儞僾儖僼傽僀儖偲僎儖僀儊乕僕偺僨乕僞(僎儖僼傽僀儖)傪奺帺偺儊僨傿傾偵曐懚偡傞丅嫟梡僐儞僺儏乕僞偵挿偔挋傔側偄傛偆偵怱偑偗偨偄丅
###########################
x 揹婥塲摦傪奐巒偡傞帪崗偑抶傟偨応崌
###########################
揹婥塲摦傪奐巒偡傞偺偑抶傟偨応崌偼丄帺摦夝愅偁傞偄偼帺摦報嶞偺愝掕傪偟側偄偱偍偔丅揹婥塲摦廔椆屻偵Gel僼傽僀儖偺傒棙梡幰偺僼僅儖僟乕傊堏摦偟偰偍偗偽丄屻擔偵夝愅傗報嶞偐傜嵞奐偱偒傞丅Gel僼傽僀儖傪暿偺僐儞僺儏乕僞偱夝愅偡傞偙偲傕壜擻偱偁傞丅
丂儊乕僇乕偺儅僯儏傾儖偱偼侾俉帪娫偺塲摦傪慐傔偰偄傞偑丄15帪娫埲忋偵挿帪娫揹婥塲摦偟偰傕丄僔僌僫儖偼摼傜傟傞偑暘棧擻偺尷奅傪挻偊偰偄傞偺偱幚幙揑偵柍懯偲側傞偙偲偑懡偄丅15帪娫偺僎儖揹婥塲摦偱800墫婎挿掱搙傑偱偺僔僌僫儖専弌偑峴傢傟傞丅僨乕僞偺棙梡栚揑偵墳偠偰嵟彫尷偺揹婥塲摦帪娫偵梷偊傞丅
帺摦夝愅傗報嶞偺娫偵丄僎儖斅傪偼偢偟婡夿晹暘偺怈偒庢傝傗儀僢僼傽乕憛偺愻忩傪偡傟偽懸偪帪娫傪桳岠棙梡偱偒傞丅
婡夿偺憖嶌儊僯儏乕傊栠傞
儊僯儏乕傊栠傞
#################
僨乕僞偺廋惓
#################
僨乕僞偺僠僃僢僋偲廋惓偼僋儘儅僩僌儔儉傪報嶞偟偰峴偄丄偙傟傪婎偵奺帺偺僐儞僺儏乕僞忋偱EditView僾儘僌儔儉傪梡偄偰僨乕僞僼傽僀儖偵廋惓傪壛偊傞丅
僋儘儅僩僌儔儉偺僾儔僀儅乕嬤朤偱偼傾乕僥僼傽僋僩偺僔僌僫儖偑懡偄偺偱墫婎傪嶍彍偡傞丅
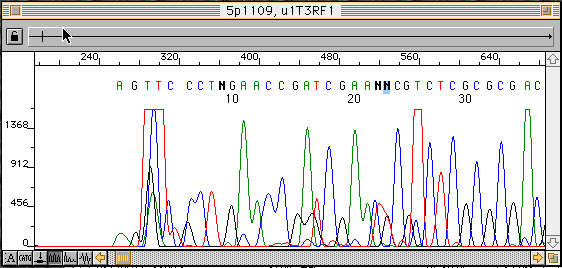
傑偨丄600墫婎傪墇偊偰丄摨偠墫婎偑楢懕偟偨晹暘側偳偱僺乕僋偑暘偐傟偵偔偔側傞偺偱岆傝偑懡偄丅埲崀傪嶍彍偡傞丅撉傒偲傝奐巒埵抲傪偢傜偟偰庢偭偨暋悢僨乕僞傪廳偹崌傢偣傞偨傔偵丄懡彮僺乕僋偺暘棧偺埆偔側偭偨晹暘傪巆偟偰偍偔偙偲傕偁傞丅
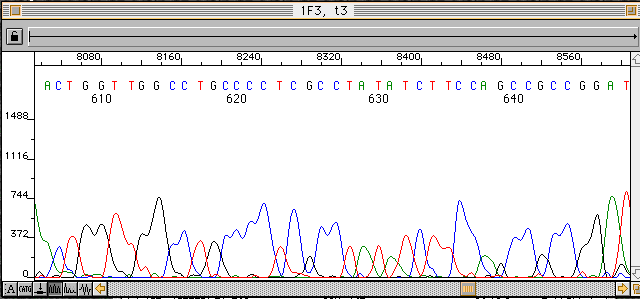
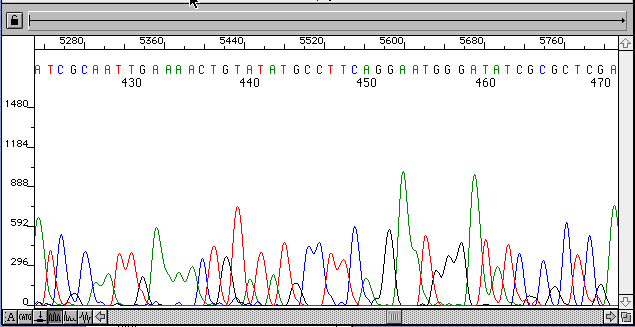
EditView偁傞偄偼Analysis僾儘僌儔儉偱僨乕僞僼傽僀儖偵廋惓傪壛偊傞偲暲楍偺墫婎攝楍偺傒偺僥僉僗僩僨乕僞傕廋惓偝傟傞丅
儊僯儏乕傊栠傞
#################
僨乕僞偺摑崌壔
#################
________________________________________________________
侾偮偺堚揱巕傗cDNA偱傕侾僒儞僾儖偺墫婎攝楍僨乕僞偺傒偱偼慡攝楍傪撉傓偙偲偼偱偒側偄偺偱丄悢屄偐傜悢廫屄偺僨乕僞傪偮側偓崌傢偣傞昁梫偑偁傞丅
憡曗嵔DNA偺攝楍偲偮側偄偩傝懳斾偝偣傞昁梫傕偁傞丅偙偙偱梡偄傞僜僼僩僂僃傾偵偮偄偰傆傟偰偍偔丅
________________________________________________________
廬棃偐傜偁傞墫婎攝楍夝愅僜僼僩丄椺偊偽Genetyx(僜僼僩僂僃傾奐敪)偱偼丄墫婎攝楍僨乕僞乮僥僉僗僩乯傪婎偵攝楍偺寢崌摑崌壔傪峴偆婡擻偑旛傢偭偰偄傞傕偺傕偁傞偺偱丄攝楍僨乕僞偺摑崌曇廤偵棙梡偱偒傞丅
曇廤偟傛偆偲偡傞墫婎攝楍娫偱僨乕僞偵柕弬偑惗偠傞偙偲傕偟偽偟偽偁傞丅惗僨乕僞偵嬤偄忣曬傪婎偵偳偪傜偺僨乕僞偑惓偟偄偺偐敾掕偡傞昁梫偑偱偰偔傞丅丂戝偒偄堚揱巕傗僆儁儘儞偺暘愅乮摉慠偵崅偄惛搙偱峴傢傟傞乯偱偼攝楍寛掕偵愯傔傞専徹偺妱崌偑崅偔側傝丄僨乕僞悢傕懡偄偺偱暋嶨偱偁傞丅偙傟傜偺嶌嬈傪岠棪揑偵峴偆偨傔偺僜僼僩僂僃傾偑奐敪偝傟偰偄傞丅
丂丂SeqEd偲AutoAssembler偼ABI幮偑奐敪偟偨僾儘僌儔儉偱丄摨幮偺僔乕働儞僒乕偺僨乕僞傪暋悢摨帪偵昞帵懳斾壜擻偱僨乕僞僼傽僀儖偵廋惓傪壛偊傞偙偲傕偱偒傞丅DNA sequencher(Gene Code Corp.)偱偼丄僨乕僞僼傽僀儖偵廋惓傪壛偊傞偙偲偼偱偒側偄偑,暋悢幮偺僔乕働儞僒乕偺僨乕僞傪暘愅曇廤偱偒傞揰偱桪傟偰偄傞丅偄偢傟傕崅壙側僜僼僩偱偁傞偑丄曇廤帪偺尒棊偲偟傗僄儔乕偼墫婎攝楍偺惛搙偵戝偒偔塭嬁偡傞偺偱摫擖傪慐傔偨偄丅
1) Maxam A.M., Gilbert W. (1977) Proc. Natl. Acad. Sci. U.S.A. 74:560-564.
2) Sanger F., Nicklen S., Coulson A.R. (1977) Proc. Natl. Acad. Sci. U.S.A. 74:5463-5467.
3) Smith L.M., Sanders J.Z., Kaiser R.J., Hughes P., Dodd C., Connell C.R., Heiner C., Kent S.B.H., Hood L.E. (1986) Nature 321:674-679.
4) Ansorge W., Sproat B.S., Stegemann J., Schwager C. (1986) J. Biochem. Biophys. Methods 13:315-323.